rgManQQtest1
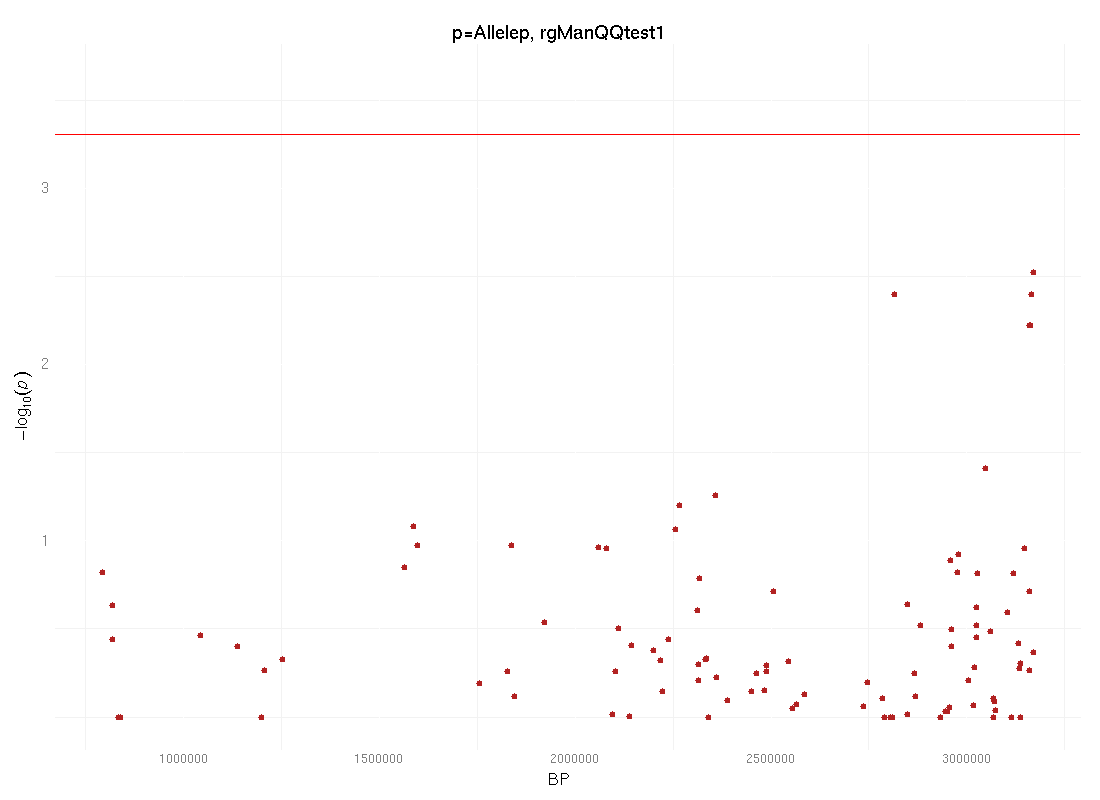 (Click to download image Allelep_manhattan.png) |
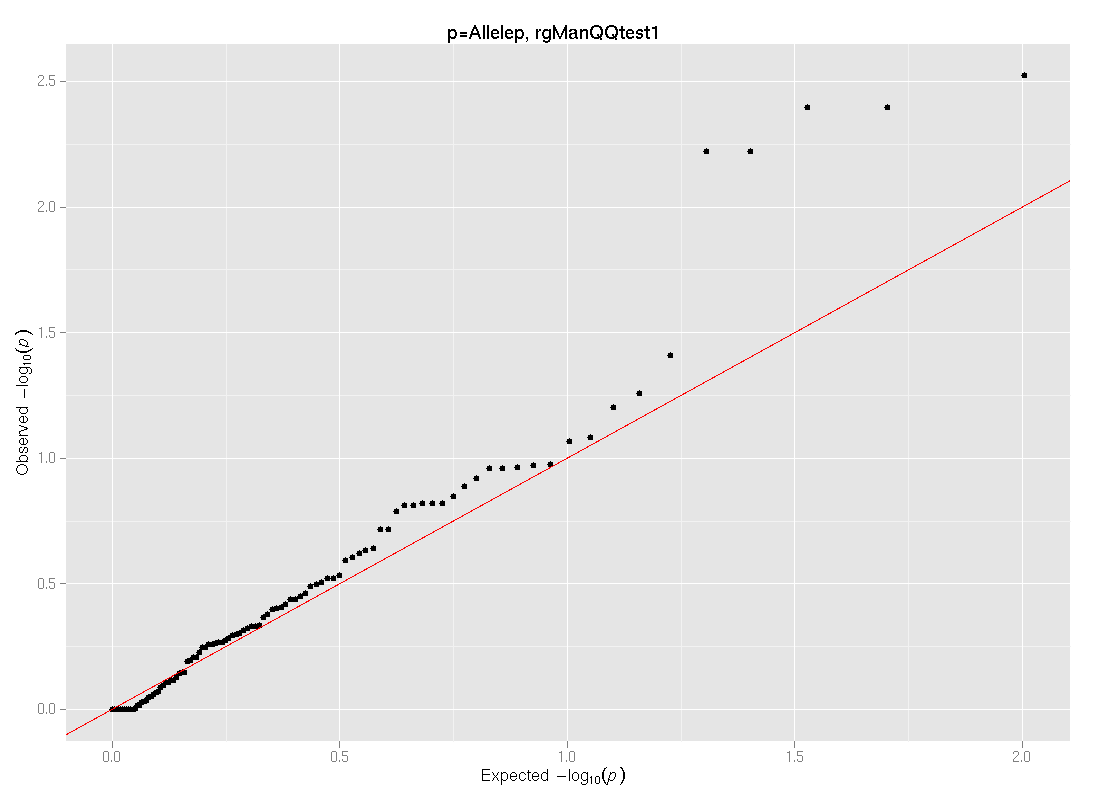 (Click to download image Allelep_qqplot.png) |
| rgManQQtest1.R |
| rgManQQtest1.R.log |
R log follows below
Loading required package: reshape
Loading required package: plyr
Attaching package: 'reshape'
The following object(s) are masked from 'package:plyr':
round_any
Loading required package: grid
Loading required package: proto
[1] "### 101 values read from /tmp/rgManQQtempcWfFkc read - now running plots"
[1] "## qqplot on Allelep done"
[1] "## manhattan on Allelep starting 1 2 3"
[1] "## manhattan plot on Allelep done"
## R script=
# license not stated so I'm assuming LGPL is ok for my derived work?
# generalised so 3 core fields passed as parameters ross lazarus March 24 2010 for rgenetics
# Originally created as qqman with the following
# attribution:
#--------------
# Stephen Turner
# http://StephenTurner.us/
# http://GettingGeneticsDone.blogspot.com/
# Last updated: Tuesday, December 22, 2009
# R code for making manhattan plots and QQ plots from plink output files.
# With GWAS data this can take a lot of memory. Recommended for use on
# 64bit machines only, for now.
#
library(ggplot2)
coloursTouse = c('firebrick','darkblue','goldenrod','darkgreen')
# not too fugly but need a colour expert please...
manhattan = function(chrom=NULL,offset=NULL,pvals=NULL, title=NULL, max.y="max",
suggestiveline=0, genomewide=T, size.x.labels=9, size.y.labels=10, annotate=F, SNPlist=NULL,grey=0) {
if (annotate & is.null(SNPlist)) stop("You requested annotation but provided no SNPlist!")
genomewideline=NULL # was genomewideline=-log10(5e-8)
if (genomewide) { # use bonferroni since might be only a small region?
genomewideline = -log10(0.05/length(pvals)) }
d=data.frame(CHR=chrom,BP=offset,P=pvals)
#limit to only chrs 1-23?
d=d[d$CHR %in% 1:23, ]
if ("CHR" %in% names(d) & "BP" %in% names(d) & "P" %in% names(d) ) {
d=na.omit(d)
d=d[d$P>0 & d$P<=1, ]
d$logp = -log10(d$P)
d$pos=NA
ticks=NULL
lastbase=0
chrlist = unique(d$CHR)
nchr = length(chrlist) # may be any number?
if (nchr >= 2) {
for (x in c(1:nchr)) {
i = chrlist[x] # need the chrom number - may not == index
if (x == 1) { # first time
d[d$CHR==i, ]$pos=d[d$CHR==i, ]$BP
tks = d[d$CHR==i, ]$pos[floor(length(d[d$CHR==i, ]$pos)/2)+1]
} else {
lastchr = chrlist[x-1] # previous whatever the list
lastbase=lastbase+tail(subset(d,CHR==lastchr)$BP, 1)
d[d$CHR==i, ]$pos=d[d$CHR==i, ]$BP+lastbase
tks=c(tks, d[d$CHR==i, ]$pos[floor(length(d[d$CHR==i, ]$pos)/2)+1])
}
ticklim=c(min(d$pos),max(d$pos))
xlabs = chrlist
}
} else { # nchr is 1
nticks = 10
last = max(offset)
first = min(offset)
tks = c()
t = (last-first)/nticks # units per tick
for (x in c(1:nticks)) tks = c(tks,round(x*t))
xlabs = tks
ticklim = c(first,last)
} # else
if (grey) {mycols=rep(c("gray10","gray60"),max(d$CHR))
} else {
mycols=rep(coloursTouse,max(d$CHR))
}
if (max.y=="max") maxy=ceiling(max(d$logp)) else maxy=max.y
maxy = max(maxy,1.1*genomewideline)
# if (maxy<8) maxy=8
# only makes sense if genome wide is assumed - we could have a fine mapping region?
if (annotate) d.annotate=d[as.numeric(substr(d$SNP,3,100)) %in% SNPlist, ]
if (nchr >= 2) {
manplot=qplot(pos,logp,data=d, ylab=expression(-log[10](italic(p))) , colour=factor(CHR))
manplot=manplot+scale_x_continuous(name="Chromosome", breaks=tks, labels=xlabs) }
else {
manplot=qplot(BP,logp,data=d, ylab=expression(-log[10](italic(p))) , colour=factor(CHR))
manplot=manplot+scale_x_continuous("BP") }
manplot=manplot+scale_y_continuous(limits=c(0,maxy), breaks=1:maxy, labels=1:maxy)
manplot=manplot+scale_colour_manual(value=mycols)
if (annotate) { manplot=manplot + geom_point(data=d.annotate, colour=I("green3")) }
manplot=manplot + opts(legend.position = "none")
manplot=manplot + opts(title=title)
manplot=manplot+opts(
panel.background=theme_blank(),
axis.text.x=theme_text(size=size.x.labels, colour="grey50"),
axis.text.y=theme_text(size=size.y.labels, colour="grey50"),
axis.ticks=theme_segment(colour=NA)
)
#manplot = manplot + opts(panel.grid.y.minor=theme_blank(),panel.grid.y.major=theme_blank())
#manplot = manplot + opts(panel.grid.major=theme_blank())
if (suggestiveline) manplot=manplot+geom_hline(yintercept=suggestiveline,colour="blue", alpha=I(1/3))
if (genomewideline) manplot=manplot+geom_hline(yintercept=genomewideline,colour="red")
manplot
} else {
stop("Make sure your data frame contains columns CHR, BP, and P")
}
}
qq = function(pvector, title=NULL, spartan=F) {
# Thanks to Daniel Shriner at NHGRI for providing this code for creating expected and observed values
o = -log10(sort(pvector,decreasing=F))
e = -log10( 1:length(o)/length(o) )
# you could use base graphics
# plot(e,o,pch=19,cex=0.25, xlab=expression(Expected~~-log[10](italic(p))),
# ylab=expression(Observed~~-log[10](italic(p))), xlim=c(0,max(e)), ylim=c(0,max(e)))
# lines(e,e,col="red")
#You'll need ggplot2 installed to do the rest
qq=qplot(e,o, xlim=c(0,max(e)), ylim=c(0,max(o))) + stat_abline(intercept=0,slope=1, col="red")
qq=qq+opts(title=title)
qq=qq+scale_x_continuous(name=expression(Expected~~-log[10](italic(p))))
qq=qq+scale_y_continuous(name=expression(Observed~~-log[10](italic(p))))
if (spartan) plot=plot+opts(panel.background=theme_rect(col="grey50"), panel.grid.minor=theme_blank())
qq
}
rgqqMan = function(infile="/tmp/rgManQQtempcWfFkc",chromcolumn=1, offsetcolumn=2, pvalscolumns=c(3),
title="rgManQQtest1",grey=0) {
rawd = read.table(infile,head=T,sep='\t')
dn = names(rawd)
cc = dn[chromcolumn]
oc = dn[offsetcolumn]
nams = c(cc,oc)
plen = length(rawd[,1])
doreorder=1
print(paste('###',plen,'values read from',infile,'read - now running plots',sep=' '))
if (plen > 0) {
for (pvalscolumn in pvalscolumns) {
if (pvalscolumn > 0)
{
cname = names(rawd)[pvalscolumn]
mytitle = paste('p=',cname,', ',title,sep='')
myfname = chartr(' ','_',cname)
myqqplot = qq(rawd[,pvalscolumn],title=mytitle)
ggsave(filename=paste(myfname,"qqplot.png",sep='_'),myqqplot,width=11,height=8,dpi=100)
print(paste('## qqplot on',cname,'done'))
if ((chromcolumn > 0) & (offsetcolumn > 0)) {
if (doreorder) {
rawd = rawd[do.call(order,rawd[nams]),]
# mmmf - suggested by http://onertipaday.blogspot.com/2007/08/sortingordering-dataframe-according.html
# in case not yet ordered
doreorder = 0
}
print(paste('## manhattan on',cname,'starting',chromcolumn,offsetcolumn,pvalscolumn))
mymanplot= manhattan(chrom=rawd[,chromcolumn],offset=rawd[,offsetcolumn],pvals=rawd[,pvalscolumn],title=mytitle,grey=grey)
print(paste('## manhattan plot on',cname,'done'))
ggsave(filename=paste(myfname,"manhattan.png",sep='_'),mymanplot,width=11,height=8,dpi=100)
}
else {
print(paste('chrom column =',chromcolumn,'offset column = ',offsetcolumn,
'so no Manhattan plot - supply both chromosome and offset as numerics for Manhattan plots if required'))
}
}
else {
print(paste('pvalue column =',pvalscolumn,'Cannot parse it so no plots possible'))
}
} # for pvalscolumn
} else { print('## Problem - no values available to plot - was there really a chromosome and offset column?') }
}
rgqqMan()
# execute with defaults as substituted